Diagnosing LAM
Because of its similarity to other lung diseases and because symptoms range from patient to patient, LAM can often be difficult to diagnose. A physician can run a number of tests to confirm or rule out the existence of LAM, to evaluate the extent of lung damage and other organ involvement.
High Resolution CT Scan (HRCT)
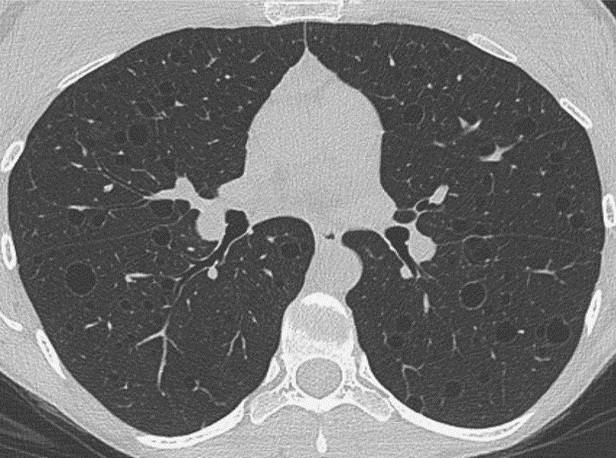
HRCT provides a detailed (2D) image of the inside of the lungs and chest. This is the most accurate and non-invasive test for diagnosing LAM, however, HRCT findings alone are not sufficient to confirm LAM. Abdominal CT scans are also used to find benign kidney tumors (called angiomyolipomas), which are present in about 40% of sporadic LAM patients.
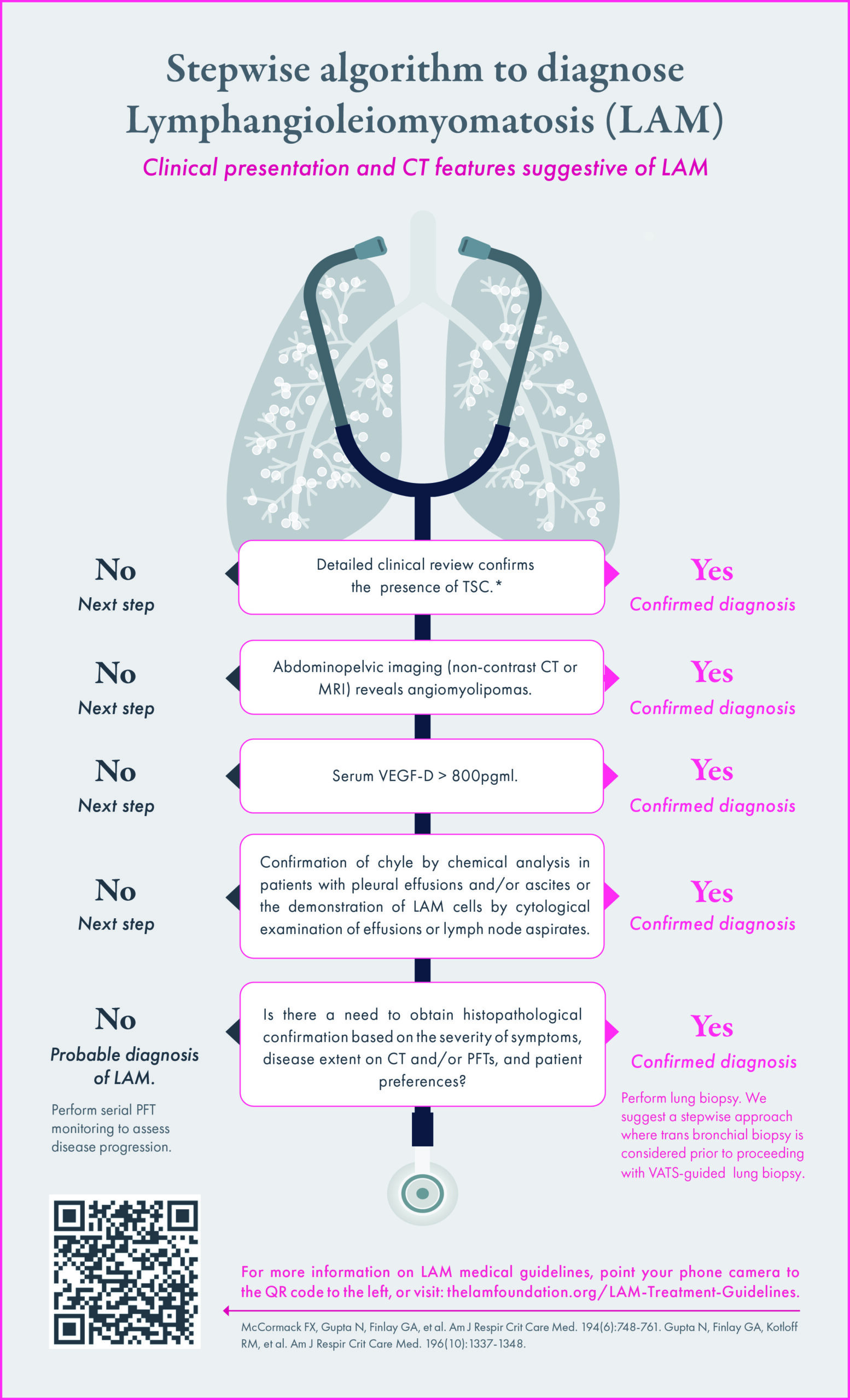
In some cases, a LAM diagnosis can be made with confidence on clinical grounds (without biopsy) in patients with typical cystic changes on HRCT of the lung plus one of the following:
- Presence of tuberous sclerosis complex
- Presence of angiomyolipoma(s)
- Serum VEGF-D > 800 pg/mL. VEGF-D is a diagnostic blood test used in conjunction with a HRCT to distinguish LAM from other cystic lung diseases; this test may also be used on women with tuberous sclerosis to screen for LAM
- Confirmation of chyle in patients with pleural effusions or acites
If none of these clinical features are present, or conclusive, a lung biopsy may be necessary to confirm the diagnosis. A lung biopsy removes small samples of lung tissue which can be examined under a microscope to look for abnormalities that may indicate a LAM diagnosis. Lung tissue can be removed in several ways:
Transbronchial Biopsy
A long, narrow, and flexible lighted tube (bronchoscope) is passed down the windpipe (trachea) and into the lungs. Pieces of lung tissue are obtained using tiny forceps. This procedure is usually done in a hospital under local anesthesia or can sometimes be done as an outpatient procedure. However, the amount of tissue obtained through this procedure may not be enough for a definitive LAM diagnosis.
Thoracoscopy, aka Video-Assisted Thoracoscopic Surgery (VATS)
Tiny incisions are made in the chest wall and a small lighted tube is passed between the ribs into the chest cavity. This allows the surgeon to view the inside of the lung and chest cavity as well as remove a small piece of lung tissue. This procedure must be done in a hospital under general anesthesia.
Open Biopsy
An incision is made in the chest wall between the ribs, allowing the surgeon’s hands into the chest cavity and tissue is removed. This procedure is done in a hospital using general anesthesia and requires a long recovery. An open biopsy should only be done if other techniques are not diagnostic or as a part of another procedure where the chest cavity must be opened.
Supplemental Testing
Chest X-Ray
A picture is taken of the heart, lungs and surrounding tissue which is usually used to show a collapsed lung or fluid build-up in the chest cavity. However, in LAM chest x-rays can be normal and is not used as the sole means of making the diagnosis.
Pulmonary Function Test (PFT)
Determines how well the lungs are working by measuring the amount (volume) of air inhaled or exhaled and how much time each breath takes (flow rates). The patient breathes through a mouthpiece into a machine called a spirometer. The spirometer records the movement of air into and out of the lungs. Although these tests are used to determine the effect LAM has on lung function, they cannot diagnose the disease.
This content was created for general informational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.